




最新资讯
咨询热线
电话:020-22091047
手机:13660676656
邮箱:2174088611@qq.com
地址:广州市番禺区南村镇陈边村金瓯东路18号金东青创园A栋601
PCR&qPCR常见33问,哪里不会点哪里
做过PCR的童鞋们都知道要想完全hold该实验、成为真正的PCR达人,并非易事。就qPCR而言,似乎其中的每一环节都可以让整个实验挂掉,也因此成为了资深科研狗们心中的痛。有时,各种假阳性、非特异性带等问题就是挥之不去、不请自来,这个时候该如何是好呢?实践出真知,小鱼就将各位前辈用经验换来的真理分享给大家。
普通PCR常见问题
Q1:PCR产物出现假阳性(即空白对照出现目的扩增产物)
Answer:
1.引物设计不合适:扩增序列与非目的扩增序列有同源性,PCR也可扩增出非靶序列的序列;靶序列太短或引物太短,可导致假阳性。此时需重新设计引物。
2.为了避免靶序列受到整个基因组或大片段的交叉污染,操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外;除了酶及不能耐高温的物质外,所有试剂及器材应高温灭菌,所有离心管及加样枪头等均应一次性使用;必要时,加样前反应管和试剂均用紫外线照射,以破坏存在的核酸;
3.为了避免靶基因受到空气中小片段核酸(与靶序列具有一定同源性)污染,可用巣式PCR方法减轻或消除。
Q2:PCR产物中出现假阴性或无扩增产物(即阳性对照中有条带,而样品则无条带)
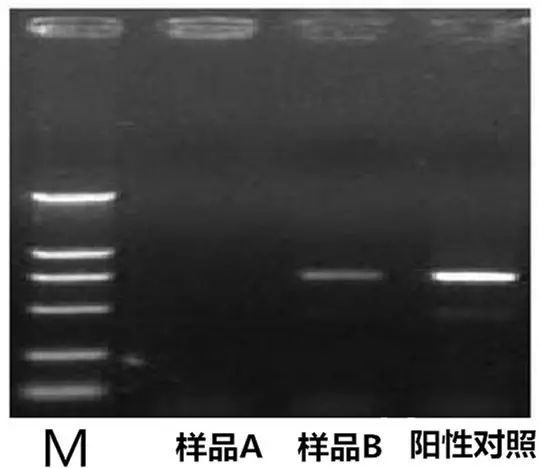
Answer:
1.条带放置时间过久,核酸被降解,最好在48h内进行电泳检测。
2.DNA模板纯度低,如含有杂蛋白质或Taq酶抑制剂,可对DNA进行再次纯化或重新用优质试剂盒提取DNA; DNA浓度太低时,可以加大模板量;对具有二级结构DNA使用较好的聚合酶;提取DNA时,避免吸入酚类试剂。
3.对设计不合理的引物进行重新设计合成;引物应高浓度小量分装保存,防止多次冻融而降解失效;检测引物OD值并进行电泳检测以确保两条引物浓度一致。
4.酶失活时,更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。
5.PCR反应条件:提高变性/退火温度;适当增加循环次数。
6.Mg2+浓度过低可影响PCR扩增产量甚至使PCR扩增失败,而Mg2+浓度过高会降低PCR的特异性,因而可适当提高Mg2+浓度。
7.如果靶序列发生突变或缺失时,也会影响引物和模板的特异性结合,产生假阴性结果。
Q3:非特异性条带扩增或者条带出现拖尾现象
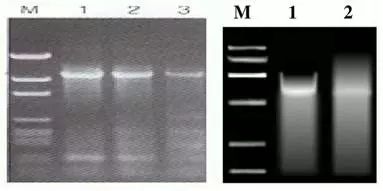
Answer:
1.当引物特异性差或引物形成二聚体时,可重新设计引物或者使用巣式PCR
2.若模板或引物浓度过高,可适当降低模板或引物浓度
3.酶量过多,则适当减少酶量
4.Mg2+浓度偏高,则降低镁离子浓度
5.退火温度偏低,适当提高退火温度或使用二阶段温度法(94℃变性,65℃左右退火与延伸)
6.循环次数过多,不仅会降低扩增效率,且会使错误掺入率增加,因此需要减少循环次数。
Q4:提高PCR特异性的策略有哪些?
Answer:
四种策略:
1.巣式PCR(Nest-PCR)可增加稀有靶序列的灵敏度;降低了扩增多个靶位点的可能性;提高PCR特异性

2.递减PCR(Touch Down PCR):前几个循环使用严谨的退火条件提高特异性;循环设在比估算的Tm高大约5℃的退火温度下开始,然后每个循环降低1-2℃,直到退火温度低于Tm 5℃ 。适合用于AFLP、DNA指纹分析等。
3.热启动PCR:抑制一种基本成分延迟DNA合成,直到PCR仪到达变性温度。如在冰上配制PCR反应液以抑制Taq酶活性,后将其置于预热的PCR仪中。
4.使用PCR增强剂:甲酰胺,DMSO,甘油,甜菜碱等可以降低熔解温度,有助于引物退火,辅助DNA聚合酶延伸通过二级结构区。但是增强剂的浓度要适当。
Q5:PCR引物设计的一般原则是什么?
Answer:
1.引物长度:15-30bp,一般为20bp左右。
2.引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。上下游引物的GC含量不能相差太大。A/T/C/G最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。
3.引物结构:避免引物3’端出现互补序列及二级结构,3’端的碱基特别是最末及倒数第二个碱基,应严格要求配对。
4.引物中酶切位点一般加在5’端,合适的酶切位点便于后续实验中的酶切分析或分子克隆。
5.引物浓度:每条引物浓度0.1-1umol或10-100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物间二聚体的形成机会。
(回复“引物”,可查看引物设计实例教程)
Q6:克隆PCR产物的最优条件是什么?
Answer:
目的片段与载体的最佳比例需依照实验来确定,一般1:1为最佳比,摩尔数比为1:8或8:1也行。连接用5ul 2X连接液, 50ng质粒DNA,1Weiss单位的T4连接酶及目的片段共10ul。室温保温1小时,或4℃过夜(可提高链接效率)。在这2种温度下,缺T-凸出端的载体会自连,产生蓝斑。
Q7:PCR产物是否需要凝胶纯化?
Answer:
如凝胶分析扩增产物中只有一条带,则无需凝胶纯化。若是有大量的引物二聚体,则需在克隆前进行凝胶纯化。
Q8:没有回收到目的片段,需要做什么对照实验?
Answer:
1.涂布未转化的感受态细胞,如有菌落,表明氨苄霉素失效,或有氨苄抗性的杂菌污染。
2.转化完整质粒,计算菌落生长数,测定转化效率。转化率=产生菌落的总数/铺板DNA总量,铺板DNA总量是转化反应所用的量除以稀释倍数。例如将1ul的质粒(1ug/ul)用于100ul感受态细胞转化。再将1ul转化后的感受态细胞稀释至1000ul后(含有10ng DNA),用100ul铺板(含有1ng DNA)。过夜培养后得到了1000个菌落。转化率=1000菌落数×103ng/铺板1ng DNA=106 cfu/ug。低于108cfu/ug,转化效率低,应重新进行细胞转化。
3.如用pGEM-T作为阳性对照,产生了超过20-40个蓝斑,表明载体失去T。可能是连接酶污染了核酸酶,需更换核酸酶。
Q9:对照实验结果好,却没有回收到目的片段,实验出了什么问题?
Answer:
1.目的片段不适合连接。因用凝胶纯化的目的片段在受到UV过度照射时会产生嘧啶二聚体,不利于连接,DNA必须重新纯化。
2.如PCR反应体系的DNA聚合酶若带有修复功能,则扩增产物末端无碱基A(该碱基是pGEM-T载体克隆所需的)。可将酶更换为Taq DNA聚合酶。
3.高度重复序列可能会不稳定,在PCR扩增中产生缺失和重排。如若目的片段高频率的产生缺失和重排,需用重组缺陷大肠杆菌菌株,如SURE细胞。
qPCR的疑难杂问
(回复“qPCR”,可查看之前相关文章)
Q10:如何提高RT-PCR反应的灵敏度与特异性?
Answer:
1.首先确定模板RNA完整性好,无DNA污染。
2.RNA模板中不应含有扩增反应抑制剂
3.为了防止模板降解,在反应体系中加入RNase抑制剂RNasin。
4.使用适量的模板RNA,模板量太多会降低特异性,太少会导致扩增不出条带或条带太弱。
5.若模板中有二级结构,可通过提高逆转录反应温度来提高扩增效果。
6.设计引物时,避免在引物3’端含有互补序列,避免形成内部发卡结构。
Q11:避免RNA降解的方法有哪些?
Answer:
1.在用来验证完整性之前先在变性胶上分析RNA
2.使用良好无污染技术分离RNA
3.将组织从动物体取出后立刻提取RNA,并将提取好的RNA反转录为cDNA进行低温保存。
Q12:RNA中含有逆转录抑制剂时,怎么处理?
Answer:
逆转录抑制剂包括:SDS、EDTA、甘油、焦磷酸钠、亚精胺和胍盐,可将对照RNA同样品混合,同对照RNA反应比较产量以检测RNA抑制剂;若对照RNA与样品混合后产量降低,则说明样品中存在逆转录抑制剂,可用70%(v/v)乙醇对RNA沉淀进行清洗,以除去抑制剂。
Q13:如何解决合成cDNA第一链合成的引物退火不充分?
Answer:
确定退火温度适合实验中所用的引物。如随机六聚体,建议在反应温度保温之前先在25℃保温10min。
Q14:如何减少RNA模板中的二级结构?
Answer:
1.将RNA和引物在不含盐及缓冲液条件下变性、退火;提高逆转录反应温度。
2.温度超过60℃时,不能使用oligo(dT)引物,选择一个在反应温度可以退火的GSP
3.RT-PCR产物的长度超过1kb时,反应温度需保持在65℃。
Q15:RNA中有DNA污染的处理方式
Answer:
1.若是基因组DNA的污染,可使用DNaseI处理RNA; 同时设置没有逆转录的对照组反应检测DNA污染。
2.若是受到外源DNA的污染,可使用抗气雾剂和UDG酶。
Q16:qPCR探针设计的一般原则有哪些?
Answer:
1.扩增片段的长度不应太大,一般小于300bp。
2.探针不能和任一引物互补,且其长度在保证特异性的前提下尽可能的短,长度不要超过30bp。
3.探针的Tm值至少比引物的Tm值高5度
4.探针如用于检测多态位点,多态位点应尽可能靠近探针中部
5.探针5’端不能是碱基G,G对荧光基团有猝灭作用。
Q17:在无反转录酶的情况下,对照RNA仍获得扩增结果
Answer:
1.体外转录时不可能将所有DNA模板消除,因而对照组会含有痕量的DNA。建议可将第一链cDNA稀释1:10、1:100、1:1000倍以消除DNA污染造成的影响。
2.可能是引物二聚体的条带。
Q18:扩增产物滞留在加样孔中
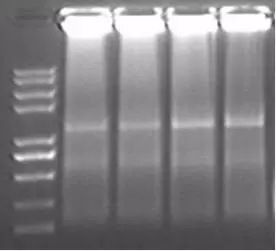
Answer:
1.可能是由于模板量过高而导致PCR结果产生了高分子量的DNA胶状物。建议将第一链cDNA浓度稀释至100倍再进行二次扩增。
2.在二次PCR时,使用的退火温度如果比引物的Tm值低5℃,可将退火温度适当增高或进行热启动以提高特异性。
Q19:无Ct信号出现
Answer:
1.反应循环参数不够,一般要在35个循环以上,但是过多的循环次数可增加背景值。
2.检测荧光信号的步骤有误。SYBRgreen法(SG法)采用的是72℃延伸时采集荧光信号,taqman法则是在退火结束或延伸结束时进行信号采集。
3.引物或探针降解,可用PAGE电泳检测其完整性,若是电泳条带呈弥散状,可考虑重新合成引物或探针。
4.模板不足或降解,则可以重新提取核酸模板。
Q20:Ct值出现过晚(Ct>38)
Answer:
1.扩增效率低,反应条件不够优化,降低退火温度,增加镁离子浓度。
2.反应成分降解或加样量不足
3.PCR产物过长,一般采用80-150bp.
Q21:标准曲线线性关系不佳
Answer:
1.加样存在误差,是样品浓度不成梯度。
2.标准品出现降解,避免反复冻融。
3.引物或者探针设计不佳。
4.模板中存在抑制物或模板浓度过高。
Q22:溶解曲线存在多个主峰
Answer:
1.引物设计不够优化。
2.引物浓度不佳,上下游引物浓度比例不一致。
3.镁离子浓度过高。
4.模板基因组的污染。
Q23:同一样品中,其中某一个荧光信号特别强。
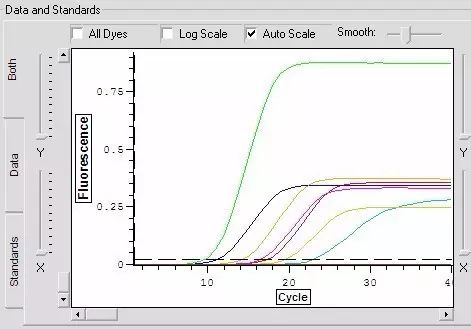
Answer:
1.试剂配制时反应液没有完全溶化,导致探针量在一管增多。
2.试剂配制时没有充分混匀致各管中各成分的量不同。
3.PCR仪热槽被荧光物质污染,需要清除热槽中的污染。
Q24:扩增曲线有一向上或向下的尖峰?
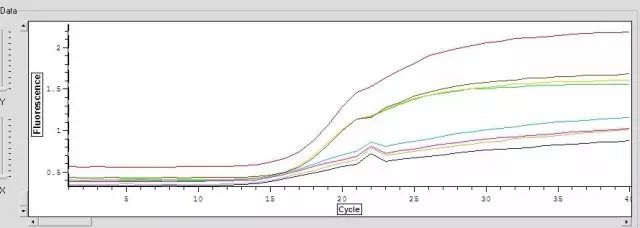
Answer:
1.反应过程中电压不稳定
2.可能在20个循环左右时,仪器有停下或仪器有开盖,使光线突然增强
3.如果尖峰向下,可能是由卤素灯老化所致,这时应更换。
Q25:部分样本扩增效率过低?
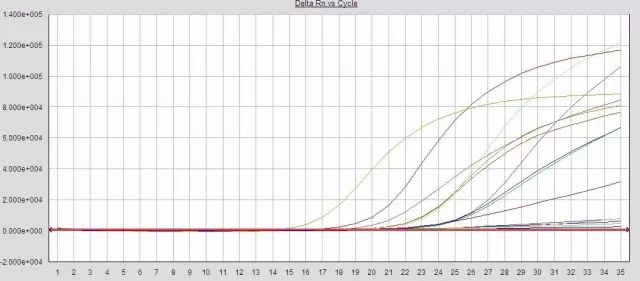
Answer:
1.提取液残留,一定程度抑制了PCR反应
2.反应液未严格取量混匀或分装不均匀
3.试剂失效
Q26:阴性对照或空白对照翘尾
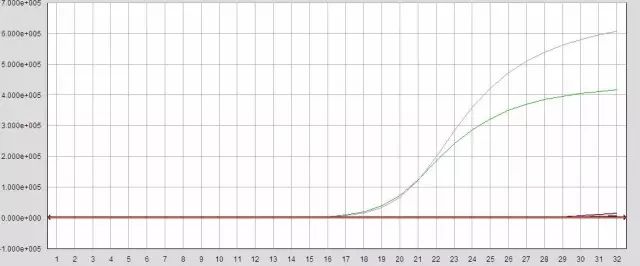
Answer:
1.模板提取环境或操作过程有污染
2.配液过程存在污染
Q27:直线型扩增曲线
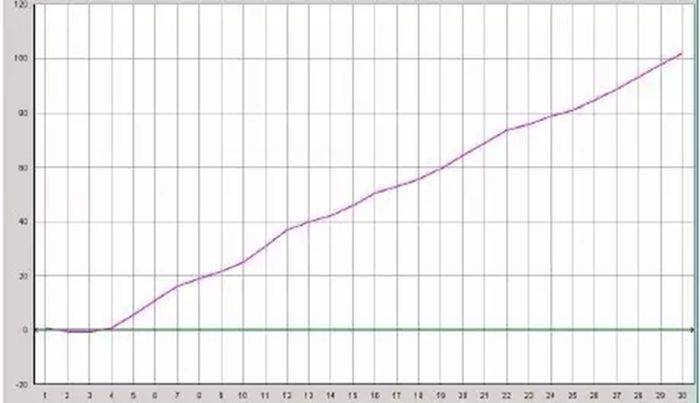
Answer:
1.探针部分降解:一般稀释的探针可在4°C保存至少3个月,探针的反复冻融或导致降解;或者探针在光线下暴露时间太长了。
2.反应液中有PCR抑制物。
Q28:没有扩增曲线
Answer:
1.PCR参数设置错误,在设计循环参数时将荧光信号读取时间设在反应的第一步,即stage1阶段
2.电脑设定了自动休眠
Q29:基线下滑
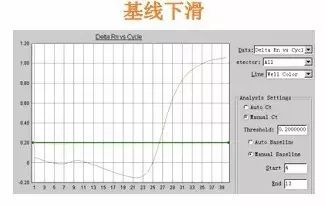
Answer:
基线选取范围不对,可试着将基线范围改大一些,这一问题常因试剂质量所致。
Q30:扩增曲线断裂

Answer:
基线选取范围不对,基线终点大于Ct值,这通常是由于模板DNA浓度过高所致,因Ct值<15,而基线范围仍取3-15,其中包含部分扩增信号,导致标准差偏大,阈值过高,解决办法:减少基线终点至Ct前4个循环,重新分析数据。
Q31:样品浓度跨度过大
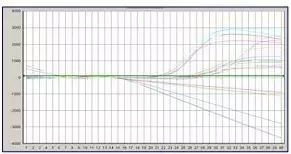
Answer:
样品浓度过高,导致阳性样品扩增曲线在后面循环中呈一向下的直线,其解决方法同“扩增曲线断裂”。
Q32:山坡形曲线
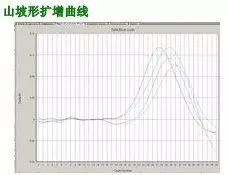
Answer:
常因反应管封口不严,至反应液蒸发所致
